1. In the Kimura 2-parameter model of sequence evolution…
a. all nucleotide substitutions are equally likely, and all nucleotides occur with equal frequencies
b. transitions and transversions occur at different rates, and all nucleotides occur with equal frequencies.
c. transitions and transversions occur at different rates, and each nucleotide can occur with different frequencies.
d. each nucleotide substitution can occur at a different rate, and each nucleotide can occur with different frequencies.
2. If a single organism contains two loci encoding homologous genes as the result of a gene duplication, the sequences encoded at these loci are said to be:
a. Orthologous
b. Metalogous
c. Paralogous
d. Xenologous
3. What characteristics of protein-coding regions are (potentially) useful for detecting these sequences among genomic sequence?
a. Patterns of base-compositional bias.
b. Lower frequency of stop codons in one reading frame than would be expected under a random model.
c. Fragments of sequence that closely resemble those of known protein-coding genes.
d. All of the above
4. Assume that the trees shown represent the phylogenies determined from phylogenetic analysis of sequences of genes encoding Tubulin and ATPase. You will note that the two analyses have produced mutually incompatible trees. Which of the following is a plausible explanation for this observation?
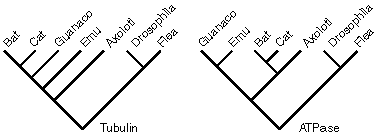
a. The data and/or phylogenetic methods used were not adequate to generate a fully resolved tree in at least one of the analyses.
b. There has been a horizontal gene transfer of the Axolotl ATPase gene to an ancestor of the Bat/Cat lineage
c. The investigators have unintentionally sequenced paralogous genes.
d. All of the above.
5. Which explanation would you favor if all of the bootstrap values on the tree were very poor (less than 30%)?
a. The data and/or phylogenetic methods used were not adequate to generate a fully resolved tree in at least one of the analyses.
b. There has been a horizontal gene transfer of the Axolotl ATPase gene to an ancestor of the Bat/Cat lineage
c. The investigators have unintentionally sequenced paralogous genes.
d. This does not help distinguish among the possibilities.
6. Which explanation would you favor if the bootstrap values on the trees were high (above 90%), and the Bat Genome Project identified a second Bat ATPase sequence that grouped with the Guanaco sequence in phylogenetic analyses?
a. The data and/or phylogenetic methods used were not adequate to generate a fully resolved tree in at least one of the analyses.
b. There has been a horizontal gene transfer of the Axolotl ATPase gene to an ancestor of the Bat/Cat lineage
c. The investigators have unintentionally sequenced paralogous genes.
d. This does not help distinguish among the possibilities.
Questions 7-10: Fill in the Blank. Provide the single most appropriate word or phrase. 5 pts. each.
7. In parsimony analysis, the _________________ that requires the smallest number of character-state transitions is preferred.
8. Maximum likelihood, minimum evolution, and maximum parsimony are ____________________ criteria.
9. GenBank is the comprehensive database in the USA for published ___________ sequences.
10. The GenBank ___________________ is a unique identifier for a sequence that is permanently associated with that sequence.
Questions 11-15: Short Answer. Provide a one or two sentence response to the question. 10 pts each.
11. What is a motif (in the context of polypeptide sequence analysis and homology determination)?
12. Define homology and distinguish it from similarity.
13. What is bioinformatics?
14. What does it mean if a region of sequence is said to be "information poor"?
15. Imagine that you are performing an analysis of some DNA sequences that you obtained by "shotgun" cloning of genomic DNA. You find a sequence that contains a long open reading frame, and use BLASTN to examine GenBank for similar sequences. BLASTN returns a peculiar group of sequences that have a short region (ca. 30 NT) of perfect match, but the rest of the sequences seem completely unrelated (see figure below). How can you explain this observation? What could you do to improve your chances of identifying sequences that are homologous to the sequence you have obtained?

Questions 16-18. Essay Questions. Provide a full response to the question. You should be able to satisfactorily answer each question with a few lines, and brevity is preferred, but be sure to completely answer each component of the question. 25 pts. each.
16. Describe how pairwise sequence alignment methods (such as the Smith-Waterman algorithm) can be extended to multiple sequence alignment. What potential problems would you anticipate with the approach you recommend?
17. Deinococcus radiodurans is a radiation-resistant bacterium that is capable of surviving exposure to enormous doses of radiation. It is known to be polyploid, i.e., it has multiple copies of each chromosome. Describe the mechanisms that confer radiation resistance to D. radiodurans, with special reference to its polyploidy and capacity for DNA repair.
18. Briefly compare and contrast maximum parsimony, distance (particularly minimum evolution), and maximum likelihood methods of phylogenetic analysis, as applied to DNA sequences. To do so, evaluate each in terms of its performance with highly divergent sequences, underlying assumptions, computational speed, and the relative efficiency with which each method takes advantage of the information contained in DNA sequences.
Question 19: Random Question to Make the Points Add Up: Provide a plausible answer. 1 pt.
19. Who are you rooting for, Celera Corp. or the publicly funded Human Genome Project? Why?
-end-