- Familiarity with the Unix family of operating systems.
- Comfort with the world wide web.
Become familiar with the blast family of alignment algorithms. This includes the empirical properties of estimating error and significance. Get a sense of the size and scope of GenBank and study how different BLAST variants behave when used to search over GenBank.
By the end of 2002 the GenBank database had over 28x109 base
pairs of DNA sequence data. Some of this has been annotated, but much of
it either has no or even worse; incorrect annotation. Given this
tremendous quantity of uncharacterized information, how can one find
sequences of interest!?
One common method is to look for sequences which are similar to a known
sequence. Several search algorithms have been developed that can search a
large database for sequences similar to a query sequence.
Among the most important algorithms currently used to search sequence databases (as of 2003) are a family of algorithms based on BLAST, the "Basic Local Alignment Search Tool." BLAST performs particularly well with protein-coding sequences. BLAST is much faster but potentially less sensitive than an older algorithm called FASTA which is also used to search large sets of data. FASTA may indeed prove more effective with non protein-coding DNA sequences.
Searching a large sequence database is a difficult problem because there are many ways the query sequence might align with sequences in the database. In order to expedite this process, BLAST looks for small regions of perfect match between the query and target sequences, and then examines the sequence that adjoins these regions to see if there is a longer stretch that matches perfectly.
Consider the following DNA sequence:
ATTTGGAGCATCATGCCTGCAAACTCCGAGAAGGAGCACCTCTCCATCGT GATTTGCGGCCATGTCGACAGTGGCAAGAGCACCACAACAGGGCGGCTC TCTTCGAGCTCGGTGGCCTTCCAGAGCGCGAACTTGACAAGCTGAAGCA GAGGCTGAGCGTCTTGGGAAAGGTTCTTTCGCCTTTGCATTCTACATGGA CCGGCAGAAGGAGGAGCGTGAGCGTGGGGTGACCATCGCTTGCACCACG AGGAGTTCTACACCGAGAAGTGGCACTACACAATCATTGATGCACCGGGC CACCGTGATTTCATCAAGAACATGATCACGGGTGCATCCCAGGCTGATGT CGCACTCATCATGGTTCCCGCAGACGGAAACTTCACGACAGCAATCGCCA AGGGCAACCACAAGGCGGGGGAAATCCAGGGCCAGACCAGGCAGCATTCC CGGCTCATCAACTTGCTTGGCGTGAAGCAGATCTGCATTGGCGTGAACAA GATGGACTGCGACACGGCGGCATACAAGCAGGCCCGTTATGATGAGATTG CAAATGAGATGAAGAGCATGCTCGTGAANGTCGGGTGGAAGAAGGACTTT ATTCGAGAAAACACACCCGTGATGCCCATCT
This DNA sequence was obtained by arbitrary screening of a cDNA library. We would like to learn more about the sequence. One easy way of gaining insight into a sequence is to find out whether or not it resembles seqeunces previously characterized. BLAST is capable of comparing the sequence to the GenBank database maintained by NCBI (the National Center for Biotechnology Information, a branch of the NIH National Library of Medicine). We will use the sequence above as a query sequence, and use BLAST to compare the query sequence to the GenBank database. The actual analysis will be run on a massively parallel supercomputer operated by NCBI as a service to the research community. There are several ways to submit searches to the blast server; we will start with the web interface.
Note! It is essential that you understand how different computers interact to perform the analyses that you are carrying out. When you use a web browser to connect to a web site, you are initiating a host/client interaction. Your desktop computer is the client, the computer that is running the web host software is the host. In this case you will be running a computationally intensive task on the host computer, so the apparent speed with which the analysis runs will be a function of the load on the host computer (among other factors).
First, copy the sequence. Then go to the NCBI web site:
http://www.ncbi.nlm.nih.gov/,
follow the link for BLAST on the NCBI home page, and then the link for
Nucleotide-nucleotide BLAST [blastn].
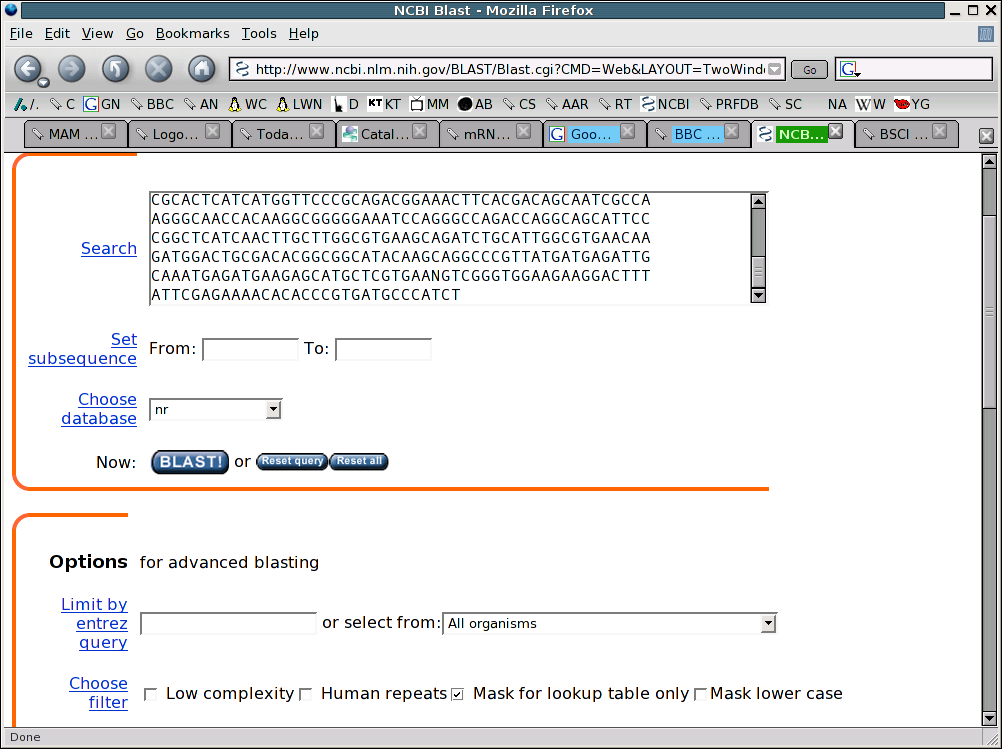
In the space provided, paste the sequence and then click on the button that says
BLAST! Two common choices for filters are chosen at the bottom of the
above screenshot. The first turns off the removal of repetitive sequence while
the second turns filtering on only for the first portion of the blast search.
The page will be replaced with a page called "formatting BLAST." Notice that it provides you with a blast ID number, an estimate of how long it will take for the results to be returned, and some formatting options.
While you are waiting for your blast results to be returned, open up another browser window and expore the NCBI home page. You should read the blast overview: http://www.ncbi.nlm.nih.gov/BLAST/blast_overview.html, and other information linked to the blast page.
Return to the 'Formatting BLAST" page and click on the FORMAT button. The results of your search will be displayed. There is information on how to cite this analysis in scientific publications and on the nature of your search, followed by a set of colored lines that illustrate the results of the search, and then text describing the results of the search, and below that more text showing examples of the best matches.
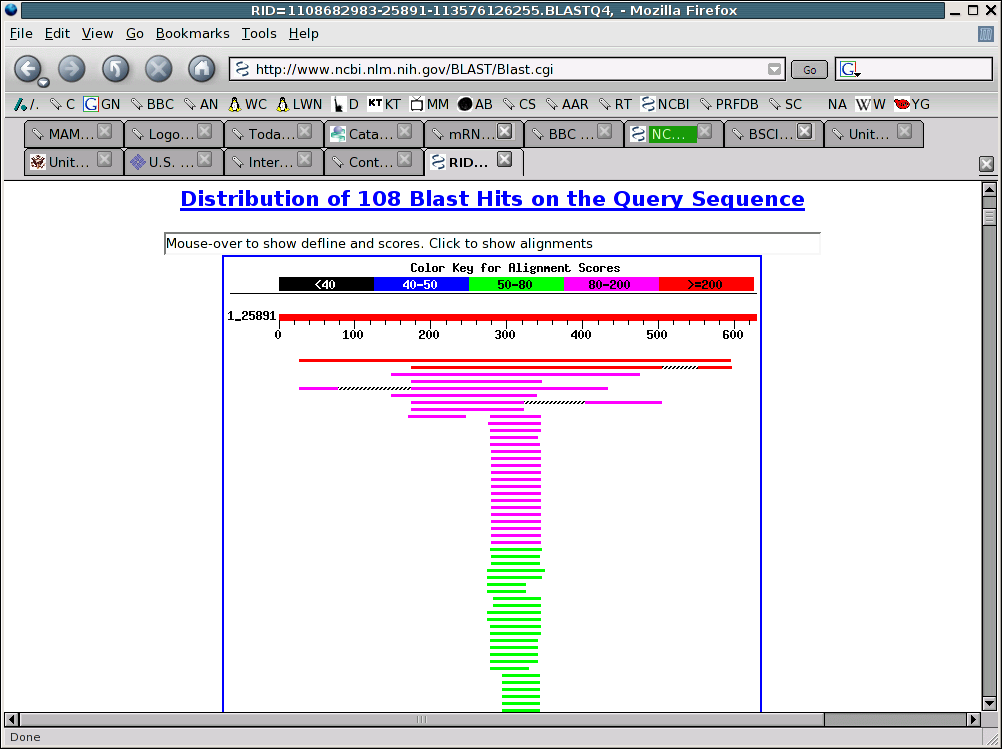
Mouse over the colored lines and notice how the display changes. Look at how this information correlates with the text further down the page, and notice that there are links to the sequences which the query sequence matched. Take some time here and try to look at all of the features on this web page.
Some questions about the test sequence:
- What inferences about this sequence can you make from this information?
- What is the identity of the sequence?
- What gene do you think it encodes?
- What organism do you think it comes from?
- How reliable do you think this inference is? Why?
Take a moment to read over the bit score, e-value, and each of the individual matches. Follow some of the links provided in order to get a sense of the web of information.
Recall that the sequence was from a cDNA library. That means that it is probably a protein-coding sequence. Blast is more sensitive to subtle patterns in amino acid sequences than in nucleotide sequences, so it can be helpful to try a search that takes advantage of the information that this is a protein coding sequence. Furthermore, we do not know the reading frame of this sequence, thus we will want to search a translation of the sequence in all six frames against a protein database.
Because you are working with a nucleotide sequence, you will need to perform a translated search. Return to the BLAST home page and under Translated BLAST Searches select Nucleotide query - Protein db [blastx].
Notice that there are a number of other options you can select, but do
not change them.
Submit the search request, and examine more of the NCBI site while waiting.
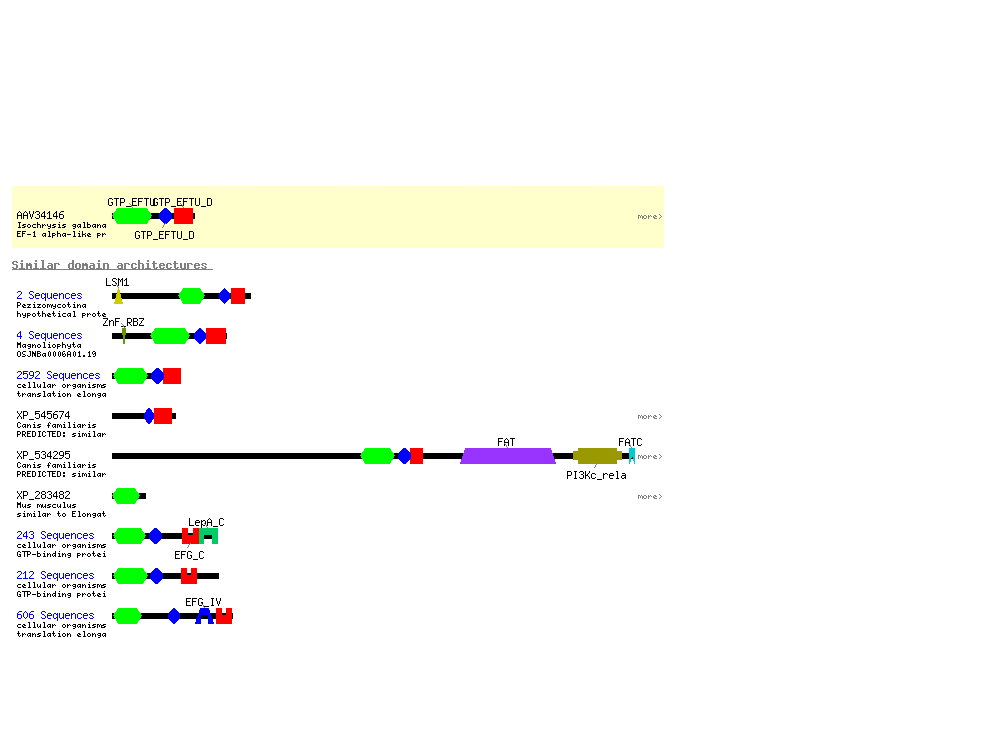
Try to find where the following images came from (a hint: they both came no
more than 3 links directly from our previous nucleotide search:

Note: Blast searches submitted to via the web site are submitted to a queue; they are given a priority that is a function of the number of searches submitted per user at the same time. Thus, if you submit a series of searches from the same computer, each search will take progressively longer. If you want to submit multiple searches it is best not to use the web interface to submit searches, instead use blastcl3 for multiple searches. This document will discuss blastcl3 momentarily.
Some questions regarding the translated blast search:
- How do the results differ from the blastn search?
- What inferences can you make from the different results in the two searches
- What is the most probable identity of the sequence?
- What gene do you think it encodes?
- What organism do you think it comes from?
- How reliable do you think this inference is? Why?
- Why do nucleotide and amino acid searches behave very differently?
- How do these two types of data differ in the way in which they carry information?
- What percent sequence identity would you expect in an alignment (without gaps) of two random DNA sequences?
Please recall that each amino acid is encoded by three nucleotides, but
that an amino acid sequence also consists of one-third the number of
characters as its corresponding nucleotide sequence. Given that information,
and the fact that the genetic code is degenerate:
What percent sequence identity would you expect in an alignment of two random
amino acid sequences?
Consider the different options, including parameters, that can be set from the BLAST page. Can you determine what effect each of these will have? Some control the way in which the BLAST results are formatted, while others control how the algorithm itself will function.
Change the word size from 11 to 7 and repeat the BLASTN search. Are the
results identical to the word size 11 search? How do the two searches differ?
What happens if you use a word size of 15?
What is the effect of changing the 'Expect' value? Download and read at least
the abstract of the paper which examines the statistics of alignments.
Additional unknown sequences are available here. Choose at least two of these and perform BLAST searches. What observations can you make about how to use BLAST most effectively?
Running BLAST from a command line interface
NCBI makes available multiple BLAST clients, blastcl3 and blastall, that can be used to launch BLAST searches from a local computer without using a web interface. Blastcl3 submits BLAST searches to ncbi for computing in a manner similar to submitting them via the web. blastall on the other hand requires a local sequence database, available at: ftp://ftp.ncbi.nlm.nih.gov/blast/db/
Code Listing |
<15:38:56>trey@sedition:> blastcl3 -p tblastx -i test.fasta TBLASTX 2.2.10 [Oct-19-2004] Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402. Query= Scaffold: 1 Nucleotide start: 1000 (1000 letters) Database: All GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS,environmental samples or phase 0, 1 or 2 HTGS sequences) 1,151,762 sequences; -309,092,411 total letters The flags provided to blastcl3 are: "-p tblastx" means to compare a translated version of my nucleotide sequence to the ncbi protein database. "-i test.fasta" specifies the input file. <15:38:59>trey@sedition:> blastcl3 -p tblastx -G 7 -E 2 -b 500 -f 9 -i input.fasta -o output.txt TBLASTX 2.2.10 [Oct-19-2004] Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402. ... This is extremely similar except it does the following: a) defines the gap penalty to 7 |
Why would having a command line interface to blast be advantageous? Looking back to our scripting exercises with the Unix shell, one might imagine writing something like:
A new B |
$ for INPUT in `ls fasta/`; do OUTDIR=outputs/ OUTFILE=$INPUT.out if ! [ -e $OUTFILE ]; then blastcl3 -p tblastx -i $FILE -o $OUTFILE fi done The above code assumes that we have multiple fasta files in a directory called 'fasta.' As we see from the -p flag for blastcl3, those fasta files are nucleotide data. Furthermore we already have a directory called 'output' into which a set of files will created containing the output from blast. Finally, check each file to ensure that the blast search has not already been performed before starting the time consuming search. |
Read the online documentation for blastcl3(1) (if your computer does not have a manual page for blastcl3, perform a search on the web to find a copy of the manual page.
Look in your sequences directory (created in exercise 1) and consider how one would perform a blast search on all of your sequences at one time. What if some files contain nucleotide information while others are amino acid? Write a script to perform a meaningful BLAST search for all of the sequences in this directory. The outputs of your blast search should go into a separate directory and all amino acid searches should be kept separate from nucleotide searches.
Perform the some of the same searches via the ncbi web interface and compare the results.
Try out different flags for a single BLAST search. Change the blast program (the -p flag), change the gap penalty, the scoring matrix, and the word size for the alignment and compare the results.
Created: Wed Sep 15 00:58:22 EDT 2004 by Charles F. Delwiche
Last modified: Mon Nov 8 15:49:44 EST 2004
by Ashton Trey Belew.