- Facility with a sequence alignment editor
- Knowledge of the dynamic programming algorithm
It is easy to imagine an extrapolation of the dynamic programming
algorithm to n dimensions where each dimension is a sequence.
This will provide an optimal alignment with one small problem; the
amount of time and amount of memory required will be proportional
to the product of the length of all aligned sequences. As you might
imagine this is not tenable for more than a very few sequences of
relatively short size. Instead clustal and pileup and friends perform
all possible pairwise alignments with the dynamic programming algorithm;
then take the relative distances between all sequences to create a
bifurcating guide tree (NOT TO BE CONFUSED OR CONSIDERED RELATED
TO A PHYLOGENETIC TREE) which is then used to arrange the order of
addition of the sequences to a progressive alignment. Experiment with
three multiple sequence alignment programs: gcg's pileup, the
opensource clustal, and the web based t-coffee. Compare/contrast their
outputs with the help of a multiple sequence alignment editor: MacClade or
potentially seaview.
Finally, explore one possible next step of evaluation by creating a hidden
markov profile and using that to search a sequence database with hmmer.
Clustal[w|x], despite its considerable age, is considered the de-facto
standard for multiple sequence alignment. It comes with both a
command-line and X interface (thus the 'w' or 'x'). For our first
experiment, look at these sequences.
For a start, take a couple of moments to learn a little about them, what
some of them are and where they come from. Once ready, open clustalw from
the command line and perform an alignment of these sequences with it,
something like this:
Code Listing: A Shell |
<22:38:53>trey@sedition:> clustalw ************************************************************** ******** CLUSTAL W (1.83) Multiple Sequence Alignments ******** ************************************************************** 1. Sequence Input From Disc 2. Multiple Alignments 3. Profile / Structure Alignments 4. Phylogenetic trees S. Execute a system command H. HELP X. EXIT (leave program) Your choice: 1 Sequences should all be in 1 file. 7 formats accepted: NBRF/PIR, EMBL/SwissProt, Pearson (Fasta), GDE, Clustal, GCG/MSF, RSF. Enter the name of the sequence file: fun.fasta Sequence format is Pearson Sequences assumed to be PROTEIN Sequence 1: NF01242439 272 aa Sequence 2: NF01242241 146 aa Sequence 3: NF00212957 386 aa Sequence 4: NF01863930 103 aa ************************************************************** ******** CLUSTAL W (1.83) Multiple Sequence Alignments ******** ************************************************************** 1. Sequence Input From Disc 2. Multiple Alignments 3. Profile / Structure Alignments 4. Phylogenetic trees S. Execute a system command H. HELP X. EXIT (leave program) Your choice: 2 ****** MULTIPLE ALIGNMENT MENU ****** 1. Do complete multiple alignment now (Slow/Accurate) 2. Produce guide tree file only 3. Do alignment using old guide tree file 4. Toggle Slow/Fast pairwise alignments = SLOW 5. Pairwise alignment parameters 6. Multiple alignment parameters 7. Reset gaps before alignment? = OFF 8. Toggle screen display = ON 9. Output format options S. Execute a system command H. HELP or press [RETURN] to go back to main menu Your choice: 1 Eventually clustal provides fun.dnd (the guide tree) and fun.aln (the alignment) |
Once clustal completes the alignment, you may open it in MacClade along
with the guide tree in order to see if there are regions which "don't
look good."
To play with clustalx, you may need to transfer your sequences to a
machine which has clustal[w|x] (locus, for example) first:
Code Listing: A Shell |
<23:06:01>trey@sedition:> scp fun.fasta abelew@locus.umiacs.umd.edu:gcg/ Note that the example shows the files were transferred into a directory named gcg on the server Enter passphrase for key '/home/trey/.ssh/id_dsa': abelew@locus.umiacs.umd.edu's password: term: Undefined variable. fun.fasta 100% 11KB 11.4KB/s 00:00 <23:06:55>trey@sedition:> ssh abelew@locus.umiacs.umd.edu Enter passphrase for key '/home/trey/.ssh/id_dsa': abelew@locus.umiacs.umd.edu's password: <23:06:58>abelew@locus:> /opt/clustalx1.83/bin/clustalx gcg/fun.fasta & (Please recall that hitting the 'tab' key while typing paths makes it possible to run this command in less than 13 keystrokes, and the & allows clustal to run in the background, causing locus to immediately return me to the prompt so one may run another command while waiting for clustal to load.) |
When clustal loads, it should look something like this:
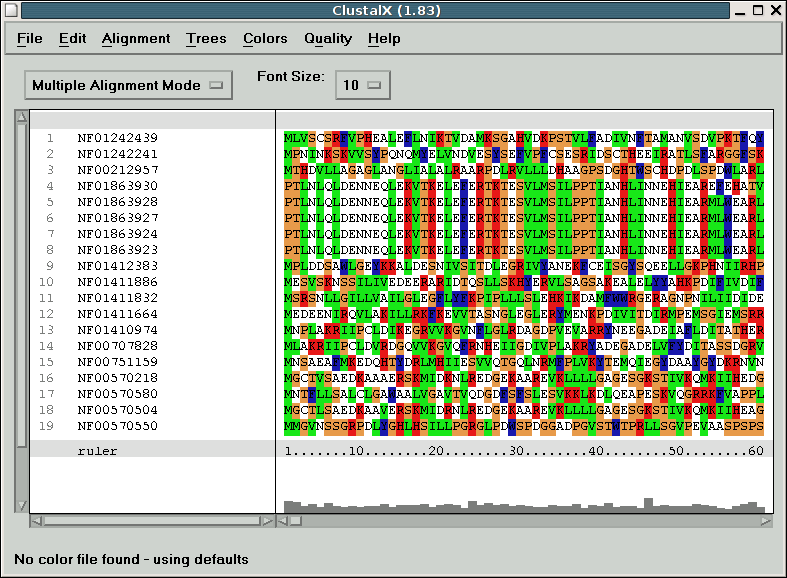
Take some time to try out the different options, change the scoring
matrix, change the output format, examine the low scoring columns, select
a subset of sequences, remove their gaps and realign _just_ those
sequences.
GCG also has some multiple sequence alignment capabilities. In order to
play with them, first examine some proteins in a family. Go to the
Protein Information Resource:
http://pir.georgetown.edu/home.shtml
and use the Search and Retrieval to do a Text search on some protein which
interests you (or use one of the many examples which have appeared in the
text book, or if you get really desperate go
here for some suggestions. Collect a group of
sequences from the pir (use the text search on a family of interest,
selected 15 interesting candidates, and used the __SAVE_OPTIONS__ dialog)
and save them to a fasta file, send it to locus, import it to gcg, and
finally use pileup upon the results:
(After collecting and sending them to locus)
Code Listing: A Shell |
<23:49:17>abelew@locus:> fromfasta fun.fasta FromFastA reformats one or more sequences from FastA format into single sequence files in GCG format. nf01242439.pep 272 aa. nf01242241.pep 146 aa. nf00212957.pep 386 aa. nf01863930.pep 103 aa. <23:50:20>abelew@locus:> pileup *.pep PileUp creates a multiple sequence alignment from a group of related sequences using progressive, pairwise alignments. It can also plot a tree showing the clustering relationships used to create the alignment. 1 nf00212957.pep 386 aa 2 nf00570218.pep 355 aa (Apparently pileup is not able to insert more than 2000 gap characters, which sounds like a lot, but it isn't; so you will quite probably have to pre-edit your sequences or cut out all sequences which are not of similar length) |
Alternatively you may want to use seqlab. Here is a session after
importing sequences of interest:
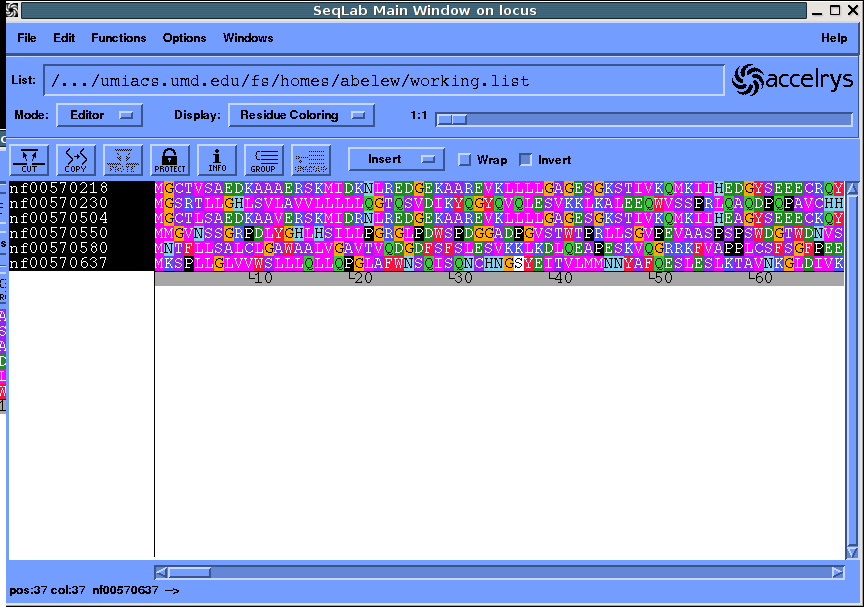
Go to the multiple sequence alignment dialog and pileup...
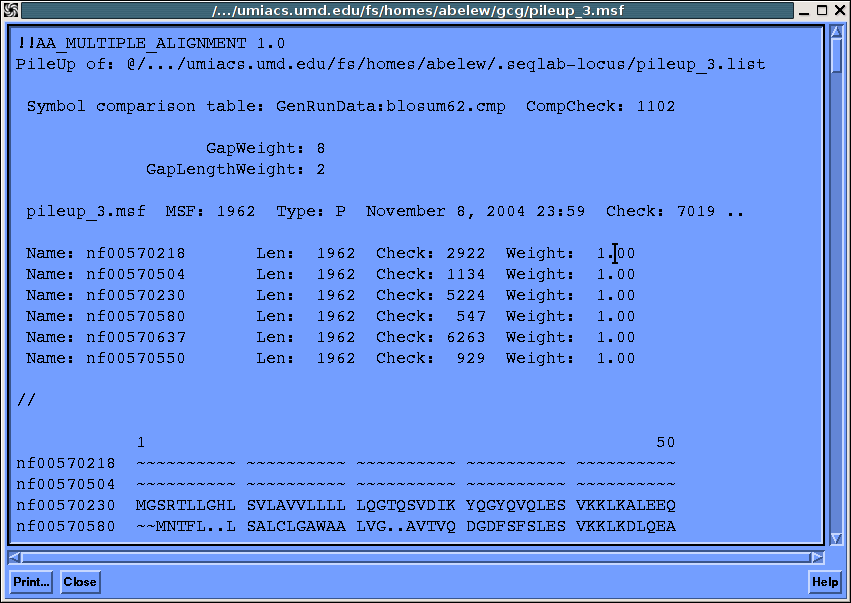
Take a few minutes and get acquainted with the gcg msa utilities.
One especially fun/interesting task you can perform is to create a
HMM profile using your multiple sequence alignment and perform a
search using it.
To collect some sequence for experimenting with T-Coffee, go to ncbi's entrez. Perform a search for ef1 alpha. Take a representative sequence and use this to perform a BLAST search. Take the sequences output from this search, download them into a fasta file. Paste this fasta file into the dialog provided at the t-coffee web site: http://www.ch.embnet.org/software/TCoffee.html to get another alignment.
Now that you have a set of alignments, it is time to examine them in
a multiple sequence alignment editor; for no machine generated
msa is perfect. Look for problems in the alignments from pileup, clustal,
and t-coffee. An alternative to using MacClade might be to use
seaview, which is not as feature rich, but is free.
Hmmer
allows one to take a complete multiple sequence alignment and create
a hidden markov model profile from it. Using this profile one may
then align more sequences to the profile, perform a search against
a secondary data source looking for other sequences which match the
profile, or modify the profile as you acquire new information.
When starting a new hmm project, the first step is to create the
initial hmmer profile:
Code Listing: A Shell |
<13:18:00>trey@sedition:> hmmbuild fun.hmm fun.aln
hmmbuild - build a hidden Markov model from an alignment
HMMER 2.3.2 (Oct 2003)
Copyright (C) 1992-2003 HHMI/Washington University School of Medicine
Freely distributed under the GNU General Public License (GPL)
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Alignment file: fun.aln
File format: Clustal
Search algorithm configuration: Multiple domain (hmmls)
Model construction strategy: MAP (gapmax hint: 0.50)
Null model used: (default)
Prior used: (default)
Sequence weighting method: G/S/C tree weights
New HMM file: fun.hmm
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Alignment: #1
Number of sequences: 22
Number of columns: 2166
Determining effective sequence number ... done. [18]
Weighting sequences heuristically ... done.
Constructing model architecture ... done.
Converting counts to probabilities ... done.
Setting model name, etc. ... done. [fun]
Constructed a profile HMM (length 504)
Average score: 266.86 bits
Minimum score: 39.29 bits
Maximum score: 572.23 bits
Std. deviation: 188.52 bits
Finalizing model configuration ... done.
Saving model to file ... done.
//
|
Given this profile one may then 'calibrate' it, which does multiple comparisons of the current profile to random sequences generated with the profile, fits the results to an Extreme value distribution, and finally resaves the profile using these new parameters.
Code Listing: A Shell |
<13:20:59>trey@sedition:> hmmcalibrate fun.hmm
hmmcalibrate -- calibrate HMM search statistics
HMMER 2.3.2 (Oct 2003)
Copyright (C) 1992-2003 HHMI/Washington University School of Medicine
Freely distributed under the GNU General Public License (GPL)
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
HMM file: fun.hmm
Length distribution mean: 325
Length distribution s.d.: 200
Number of samples: 5000
random seed: 1100024464
|
Hmmalign may be used to perform a pairwise alignment between an
individual sequence and an already existing profile.
hmmemit in turn creates random sequence which 'fits' the given
profile.
hmmsearch on the other hand does a search between a profile and a
larger dataset (somewhat like a slower but more sensitive blast),
for example:
Code Listing: A Shell |
<13:24:43>class@lfsclab05:> hmmsearch fun.hmm genpept.fasta.gz genpept.fasta.gz is a compressed copy of the genpept database from a couple of months ago hmmsearch performs a search a sequence database with a profile HMM HMMER 2.3.2 (Oct 2003) Copyright (C) 1992-2003 HHMI/Washington University School of Medicine Freely distributed under the GNU General Public License (GPL) - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - HMM file: fun.hmm [fun] Sequence database: genpept.fasta.gz per-sequence score cutoff: [none] per-domain score cutoff: [none] per-sequence Eval cutoff: <= 10 per-domain Eval cutoff: [none] - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - |
A run of hmmsearch of such a large dataset may take a couple hours, while a blast search would take approximately 2 minutes.
1. Open each of the three multiple sequence alignments in the sequence
editor of your choice and spend some time looking for the sections of each
alignment which are incorrect.
2. Go to entrez and search out papers which describe the problems
associated with performing efficient and correct multiple sequence
alignments.
3. Compare a BLAST search of a single sequence from your alignment to a
search using a multiple sequence alignment and hidden markov model.